À la découverte de nos activités dans les -omics
15/12/2021

Pour démystifier un peu ce secteur d’activité et vous offrir l’opportunité de mieux le comprendre, nos référents biostatistique et bioinformatique -omic partagent avec vous, au travers de ce premier article décrivant un pan de leur activité, un peu de l’univers dans lequel ils gravitent !
Mais au fait, qu’est-ce qu’un biomarqueur, à quoi sert-il et comment l’utiliser ?
La définition de la FDA est on ne peut plus claire à ce sujet: « un biomarqueur est une caractéristique objectivement mesurable, évaluée comme un indicateur de processus biologiques normaux, de processus pathologiques ou d’une réponse biologique à une intervention thérapeutique ». Un exemple familier pour de nombreuses personnes est l’utilisation du taux de glucose sanguin pour mesurer l’efficacité du traitement contre le diabète. Dans ce cas, le glucose est le BMK.
D’ailleurs, il existe différents types de BMKs suivis dans le cadre des études pharmaceutiques en préclinique et en clinique.
Par exemple, les BMKs macroscopiques à l’échelle d’un organe ou d’un organisme :
- les données issues de l’imagerie médicale du genou (e.g. IRM) sont par exemple des BMKs de l’arthrose,
- la pression artérielle, l’eCG permettent de suivre des évènements cardiovasculaires,
- des facteurs de risque tels que le BMI ou l’âge permettent de prévoir la susceptibilité à devenir obèse.
Mais également, les BMKs moléculaires tels que :
- les protéines sanguines dont la mesure est facilement accessible par prélèvement sanguin,
- les BMKs cellulaires (acides nucléiques, protéines, métabolites) : e.g. dans le cas de certaines mutations (causales) on peut prédire la survenue de certaines maladies.
Par ailleurs, les caractéristiques des BMKs recherchés sont principalement de trois grands ordres correspondant aux objectifs de développement produit. Les objectifs peuvent être pronostiques (en l’absence de traitement suivre l’évolution d’une maladie), prédictifs (prédire l’influence d’un traitement) ou enfin pharmacodynamiques (suivre une cinétique d’expression au cours du temps). Ces BMKs une fois identifiés permettront par exemple d’implémenter les cohortes en patients progresseurs pour une pathologie donnée, d’administrer la dose de traitement adéquate au moment le plus opportun (« 3R : Right Medication, Right Patient, Right Dose »).
Mais alors, les données « Omics » c’est quoi au juste ?
Le suffixe « Omics » tire son origine du mot sanscrit « -ome », qui désigne la complétude et la plénitude (Lederberg, McCray, 2001). La fusion avec les mots « gène » ou « transcrits » qui a donné par la suite « génome » ou « transcriptome » se réfèrent donc à l’étude d’un tout pour une certain type de molécules. En effet, la cellule exprime à chaque instant des milliers de molécules qui constituent la machinerie cellulaire aboutissant à une fonction concrète (e.g. migration, différentiation, …). Le rôle des technologies Omics est donc de capter cette information et ainsi de prendre une « image » des modifications moléculaires afin de mieux les comprendre.
Pouvez-vous me donner un exemple concret d’application ?
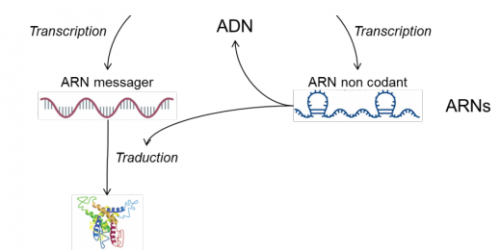
Dans toutes cellules nous retrouvons de l’ADN, transcrit ensuite en ARN messager, lui-même traduit en protéines (figure 1). Une des méthodes les plus actuelle pour évaluer la nature et l’abondance des molécules d’ADN ou d’ARN est le séquençage d’acides nucléiques (NGS : Next Generation Sequencing).
Son principe est simple : les molécules sont extraites de pools cellulaires, elles sont ensuite fragmentées et analysées via une machine appelée séquenceur. Cet appareil lit des millions de suites de nucléotides (A, T, C, G) appelés lectures (« Reads » en anglais). Chaque lecture correspond à une partie d’une séquence plus longue. L’objectif du séquençage d’ARN est d’évaluer le niveau d’expression des gènes tandis que le séquençage d’ADN permet d’identifier des modifications de la séquence génétique
La première étape est aux mains du bioinformaticien qui vérifie la validité des millions de lectures obtenues. Cette étape est très importante pour identifier les erreurs de séquençage et il existe des logiciels spécialisés qui permettent d’évaluer les paramètres de qualité des données au cours du préprocessing.
La seconde étape s’appelle l’alignement (« mapping » en anglais). Chaque fragment est comparé au génome de référence afin d’identifier sa localisation. Une alternative de plus en plus utilisée, créée au départ pour les organismes non modèles (ceux pour lesquels un génome de référence n’était pas disponible), est celle de l’assemblage de novo : méthode bioinformatique in silico, elle consiste à comparer les fragments séquencés entre eux afin de reconstruire la séquence originale de la molécule investiguée.
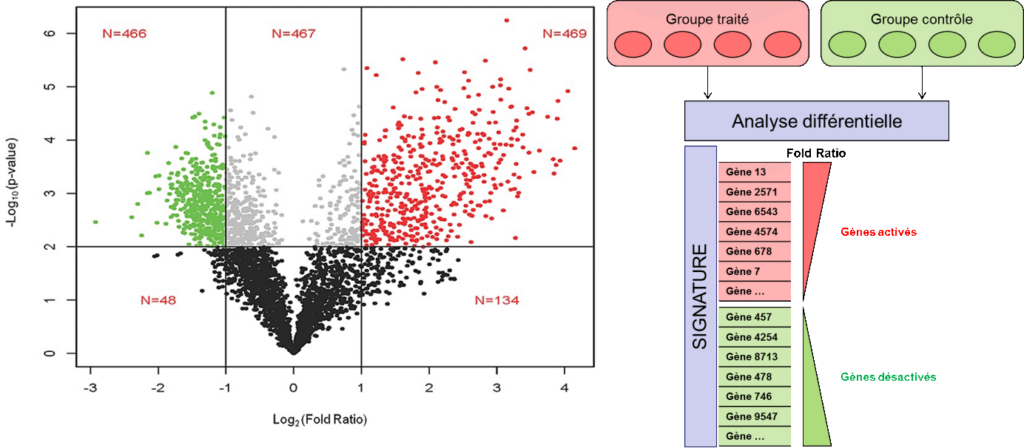
Suite à l’alignement, il est possible de comptabiliser le nombre de lectures associées à chaque transcrit. Ce résultat ne peut pas être utilisé en tant que tel à cause du biais moyenne/variance, et nécessite des transformations mathématiques/statistiques indispensables (e.g. regularized log) ou encore une modélisation de la variance adéquate (les comptages suivent des lois négatives binomiales) avant une analyse différentielle entre deux conditions d’intérêt (figure 2). Cette analyse permet notamment d’identifier des signatures divergentes entre deux états biologiques (e.g. traité vs contrôle) et constitue la base de la découverte des BMKs.
Les analyses bioinformatiques permettent également de réaliser des analyses de variation de séquence. Ces « variants » peuvent être de plusieurs types :
- des variants d’épissage (un gène est composé d’un ou plusieurs exons qui ne sont pas tous transcrits en ARN : c’est l’épissage alternatif),
- des mutations de séquence (insertions, délétions ou substitutions de nucléotides, figure 3A),
- des modifications plus globales au niveau du génome comme les translocations, observées grâce à la présence de gènes de fusion : gène hybride formé de deux gènes distants (figure 3B).
Ces variants peuvent également servir de biomarqueurs notamment dans l’analyse des cancers où la présence ou l’absence d’une certaine mutation dans les cellules tumorales permet de choisir le traitement adapté. Au final, les biostatistiques et la bioinformatique proposent aujourd’hui un ensemble d’outils mathématiques et informatiques qui constituent autant d’outils pour la découverte de nouveaux BMKs.
Le groupe
Nos engagements RSE
Conscients de notre responsabilité sociale et environnementale, nous agissons chaque jour pour faire avancer la société.
Nos actualités
Suivez toutes nos infos santé
-
 Actualité
Actualité4/05/2023
Article Test : Pourquoi le spin-off « La Reine Charlotte : un chapitre Bridgerton » est bien plus qu’une romance ?
SPIN-OFF – Outre la romance, « La Reine Charlotte : un chapitre Bridgerton » explore les que
-
 RSE
RSE7/04/2023
Clean up day : les équipes Efor donnent un coup de propre !
Dans le cadre de la semaine de la Qualité de Vie au Travail et du challenge inter-entreprise auxquelles nos équipes
-
 Article technique
Article technique3/04/2023
DM : la place des données précliniques dans la documentation technique (MDR & ISO 13485:2016)
Les dispositifs médicaux doivent être conçus et fabriqués de façon à assurer les performa