Discovering our -omics activities
15/12/2021

To demystify this area of activity and offer you the opportunity to better understand it, our biostatistics and bioinformatics -omics experts are pleased to share their sector with you through this first article describing part of their work!
What is a biomarker (BMK), what is its purpose, and how can it be used?
The FDA definition is quite clear: “a biomarker is a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or biological responses to a therapeutic intervention”. An example that is familiar to many people is the use of blood glucose levels to measure the effectiveness of a diabetes medication. In this case, glucose is the BMK.
In fact, there are various types of BMKs monitored in pre-clinical and clinical pharmaceutical studies.
For example, there are macroscopic BMKs at organ or organism level:
- data from medical imaging of the knee (e.g. MRI) are for example BMKs of osteoarthritis,
- blood pressure and ECG enable cardiovascular events to be monitored,
- risk factors such as BMI and age can predict susceptibility to obesity.
There are also molecular BMKs such as:
- blood proteins, which are easily measured by blood sampling,
- cellular BMKs (nucleic acids, proteins, metabolites): e.g. in the case of certain (causal) mutations, the occurrence of certain diseases can be predicted.
In addition, BMKs are divided into three main classes corresponding to the objectives of product development. The objectives may be prognostic (in the absence of treatment, to monitor the course of a disease), predictive (to predict the influence of a treatment) or pharmacodynamic (to monitor expression kinetics over time). Once identified, these BMKs may be used for example to implement cohorts of patients progressing for a given disease and administer the right treatment dose at the most appropriate time (3Rs: Right Medication, Right Patient, Right Dose).
Then what exactly are “omics” data?
The suffix “-omics” originates from the Sanskrit word “-ome”, which means completeness and fullness (Lederberg, McCray, 2001). Its use with words such as “gene” and “transcripts”, giving rise to “genome” and “transcriptome”, thus refers to the study of a whole for a certain type of molecule. Indeed, cells express thousands of molecules at any given moment; these constitute the cellular machinery leading to a concrete function (e.g. migration, differentiation). The role of “omics” technologies is therefore to capture this information and thus take an “image” of molecular modifications in order to better understand them.
Can you give a concrete example of an application?
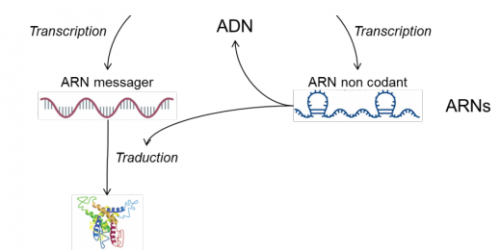
All cells contain DNA, which is then transcribed into messenger RNA, which in turn is translated into proteins (Figure 1). One of the most up-to-date methods for assessing the nature and abundance of DNA and RNA molecules is next-generation sequencing (NGS).
The principle is simple: molecules are extracted from cell pools and then fragmented and analysed using a machine called a sequencer. This machine reads millions of nucleotide sequences (A, T, C, G) called reads. Each read corresponds to part of a longer sequence. The objective of RNA sequencing is to assess the level of gene expression, whereas DNA sequencing identifies changes in the genetic sequence.
The first step is in the hands of the bioinformatician who checks the validity of the millions of reads obtained. This step is very important to identify sequencing errors and there are specialised software packages that can assess data quality parameters during pre-processing.
The second step is called mapping. Each fragment is compared with the reference genome to identify its location. An increasingly used alternative, initially created for non-model organisms (those for which a reference genome was not available), is that of de novo assembly: this is an in silico bioinformatics method that involves comparing the sequenced fragments with each other in order to reconstruct the original sequence of the molecule under investigation.
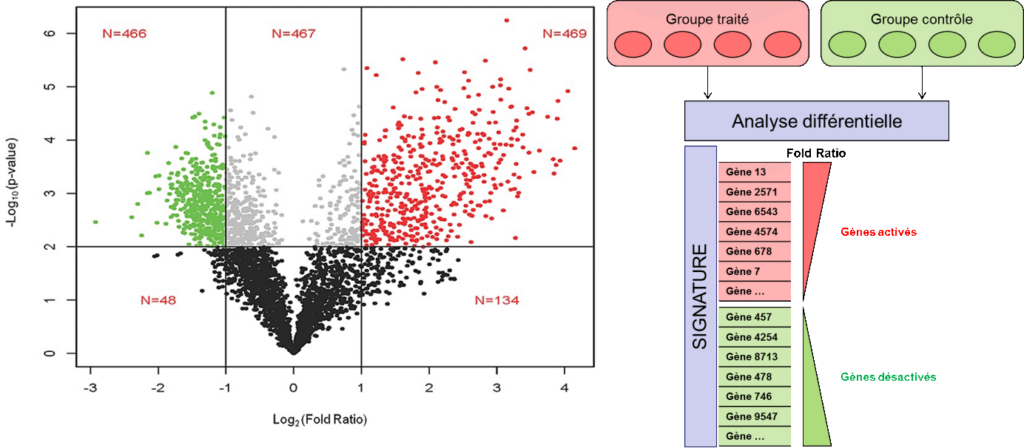
Following the mapping step, the number of reads associated with each transcript can be counted. This result cannot be used as is because of the mean/variance bias; it requires essential mathematical/statistical transformations (e.g. regularised log transformation) or adequate variance modelling (counts follow negative binomial distributions) prior to a differential analysis of two conditions of interest (Figure 2). This analysis is able to identify divergent signatures between two biological states (e.g. treated vs. control) and is the basis for discovering BMKs.
Bioinformatics analyses can also be used to analyse sequence variation. There can be several types of “variants”:
- splicing variants (a gene is composed of one or more exons that are not all transcribed into RNA: this is alternative splicing),
- sequence mutations (insertions, deletions or substitutions of nucleotides, Figure 3A),
- more general modifications at genome level such as translocations, observed thanks to the presence of fusion genes: hybrid genes formed from two distant genes (Figure 3B).
These variants can also serve as biomarkers, particularly in the analysis of cancer, where the presence or absence of a certain mutation in tumour cells enables the appropriate treatment to be chosen.
In the end, biostatistics and bioinformatics now offer a range of mathematical and computer tools for the discovery of new BMKs.
Efor group
Our CSR commitments
Aware of our social and environmental responsibility, we act every day to make a positive impact on society.
Nos actualités
Suivez toutes nos infos santé
-
 Technical articles
Technical articles3/04/2023
MD: the role of pre-clinical data in technical documentation (MDR & ISO 13485 :2016)
Medical devices shall achieve the performance intended by their manufacturer and shall be designed and manufactured in such a way
-
 CSR
CSR1/03/2023
We broke records at the Eco Sport Challenge!
On March 18th, the Lyon-based Efor teams participated in a unique inter-company challenge with great enthusiasm: picking up as man
-
 News
News11/02/2023
Efor US. is born
Something is coming …Before closing 2022 marked by growth, resilience and starting a new year rich in projects, we are laun