Changes to medical devices (MDs / IVDMDs)
20/10/2022

Once CE marked
The life of a medical device is not a long, quiet river… Who has never attended a team meeting on a change to be made to a product that has only just obtained CE marking? Amending a specification, adding a supplier, correcting a failure – all this, and much more, is part of the post-market cycle.
But can any kind of change be made? Are there rules to be followed?
Substantial change
Prior to the publication of Regulations (EU) 2017/
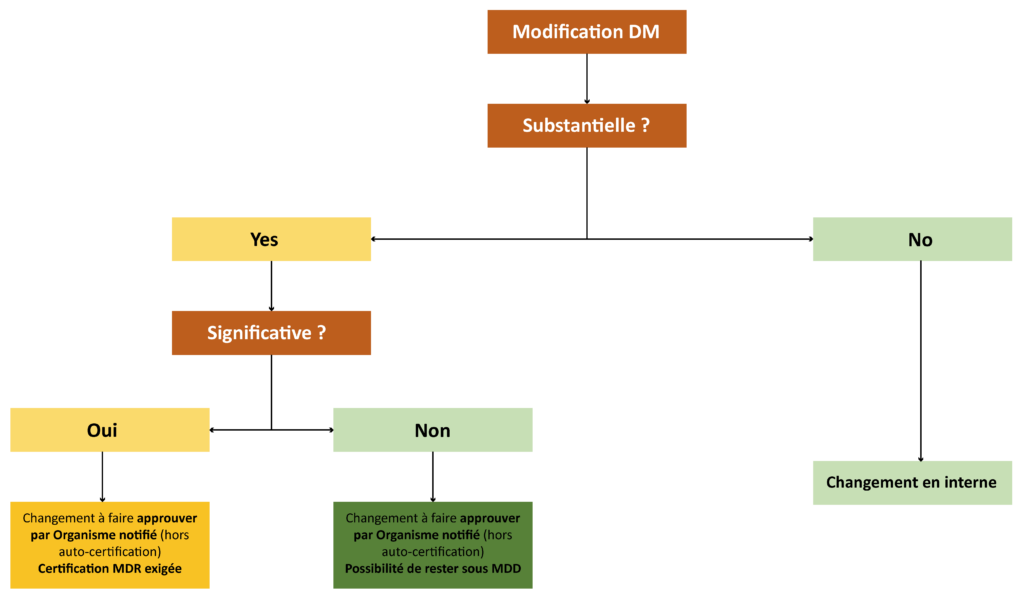
745-746, changes to medical devices (MDs and IVDMDs) certified according to Directives 93/42/EEC and 98/69/EEC were approached on an exclusively binary basis where they were considered either substantial or non-substantial (sometimes called major or minor). Put simply, a change is considered substantial when it impacts the product’s safety and/or efficacy and/or performance. Note that certain changes to the Quality Management System may also be considered substantial.
Although every change remains open to discussion and interpretation, NBOG BPG 2014-3 provides guidance on the matter enabling the appropriate conclusion to be drawn in each situation. When a change is defined as substantial, the change implementation request needs to be submitted to the notified body that conducted the compliance assessment (except for self-certified Class I or A devices). It is then necessary to demonstrate that all efforts have been made to maintain a favourable benefit-risk ratio for the patient.
Significant change
Since the entry into force of Regulations (EU) 2017/745-746, an additional consideration has emerged regarding the status of a change for devices still certified under Directives 93/42/EEC and 98/79/EEC (legacy devices). In addition to determining the substantial nature of a change, it is now necessary to assess whether it is significant or not. Initially, Articles 120 of Regulation (EU) 2017/745 and 110 of Regulation (EU) 2017/746 defined this status as being a change in the design or intended purpose. The MDCG 2020-3 guide clarified all this and included additional concepts to be taken into account for MDs (change to software or hardware or the sterilisation method). It uses decision trees to rule as to this new category of change. MDCG 2022-6 is an equivalent guide for IVDMDs.
As with a substantial change, if a change is deemed significant, the implementation request must be submitted to the notified body in charge of certification. The impact then becomes considerable. Indeed, the product’s dossier must be fully submitted in accordance with Regulations (EU) 2017/745-746 in order for the change to be processed and accepted. This will require that the MD/IVDMD be certified under the new regulations.
To summarise The following decision tree illustrates the change control process, in particular the timing of the decisions to be considered when changing a medical device:
Efor is here to assist you
Our experts in regulatory affairs, quality and other fields will be pleased to offer you support in line with your needs for all of the following topics (non-exhaustive list):
- Evaluation of your changes and definition of their status,
- Assistance implementing your changes and updating the associated technical dossiers,
- Updating of your Quality Management System to incorporate the various change control requirements according to the regulations in force.
Feel free to contact us using the contact form
Efor group
Our CSR commitments
Aware of our social and environmental responsibility, we act every day to make a positive impact on society.
Nos actualités
Suivez toutes nos infos santé
-
 Technical articles
Technical articles3/04/2023
MD: the role of pre-clinical data in technical documentation (MDR & ISO 13485 :2016)
Medical devices shall achieve the performance intended by their manufacturer and shall be designed and manufactured in such a way
-
 CSR
CSR1/03/2023
We broke records at the Eco Sport Challenge!
On March 18th, the Lyon-based Efor teams participated in a unique inter-company challenge with great enthusiasm: picking up as man
-
 News
News11/02/2023
Efor US. is born
Something is coming …Before closing 2022 marked by growth, resilience and starting a new year rich in projects, we are laun